

Introduction
In order to simulate the events in a organic solar cell or a light emitting diode (LED) there are two processes that is needed to be taken into consideration: First the absorption of an photon which, in the case of the solar cell, translates to the excitation of a molecule with a ray of light. In the LED the process in reversed with the emitting of an photon by "de-excitation" of a molecule where an excited electron falls down to a lower orbital. The next step is the actual transport or the electron (or hole) which in the case of a solar cell means that we need to transport electrons away when they have been excited in the molecule to create a currect. Again for the LED the process is reversed and the electrons (or holes) are fed into the molecule to create light with a certain wavelength. The latter process is called a charge transfer (CT) and together with the excitation they make up the foundations of light absorption and emission. These processes will be described below in a quantum mechanical and physical sound way by introducing a model including these both events. In the end this model will be compared to experimental results for verification.
Frenkel Model
The Frenkel, named after the Russian physicist Yakov Frenkel, exciton can be applied when the materials dielectric function is very small so that the Coulomb interaction between electrons and holes are very strong. This results in that the exciton size stays relative small, in the same order as a unit cell, and therefore the electron and hole resides on the same molecule with the binding energy in the order of 1.0 eV. In the Frenkel model this is the only allowed exciton and will therefore be best suited for organic materials where there are no strong covalent bindings.The starting point of the Frenkel model is a crystal in its lowest energy state where all molecules in the system are in their electronic and vibronic ground state. Within the effective mode model, a consequence of BO-approximation, it is possible to factorize the electronic ground state of the crystalline phase into the product of the ground state wave function of the constituting molecule. The operation of the Frenkel exciton operator on a system in its ground state results in a factorization of the excited state molecules wave function:


where the

Fourier Transform
As the system is periodic we can perform a total Fourier transformation into wave-vector representation to be able to include all molecules in the infinite stack, the k-space operator can be written as
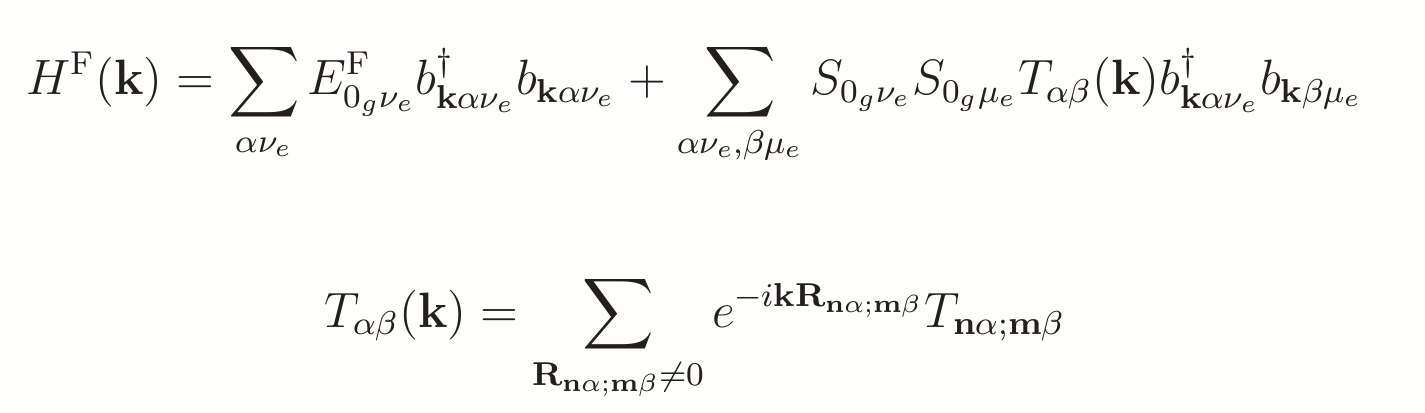
Frenkel and Charge Transport (CT) Model
The full Hamiltonian including Frenkel, CT and their mixing can summed up as


By using the same logics as before the CT Hamiltonian can be written as


For a summation of allowed transfers that are included in the model, see figure 3.2 below

To summarize one can write the Hamiltonians in their matrix representation. The hopping of CT-states are neglected here since that would imply transfers of two charge carrier simultaneously which are much less probable so we do not consider this. Under the given conditions we have periodic boundary conditions and as before trasformation via a Fourier scheme is performed into the momentum space representation.
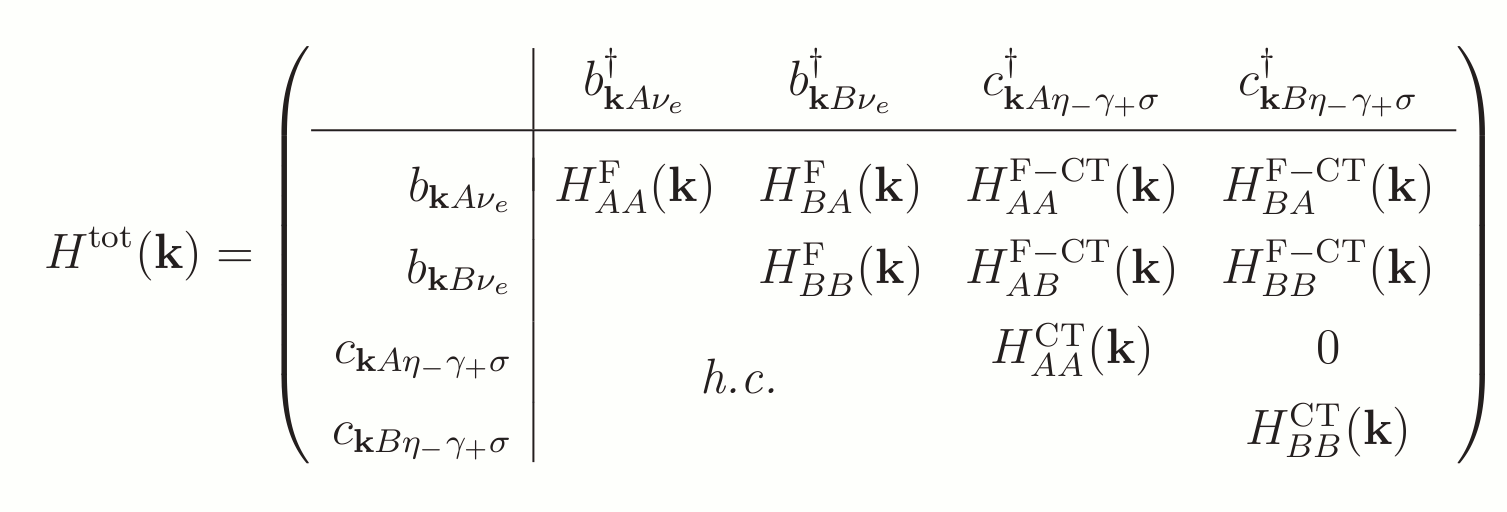
Optical Properties
The transition dipole moment for the ground state to the excited state is given by the relevent off-diagonal elements of the dipole matrix. We neglect exchange of electrons between CT dimers and the other molecules in their ground states so we get for charge transfer dipole moment. The total expression can be written as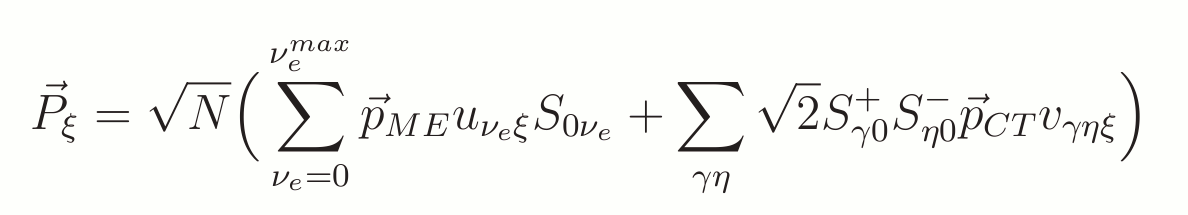

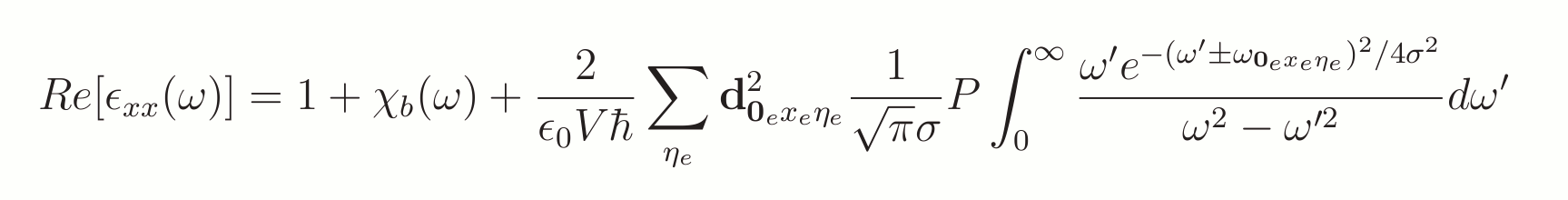
F-CT Model Results
The F-CT model optical spectra can be compared to experimental results. Below are some of the molecule spectras that were presented in my PhD. thesis. For all the molecules and their optical lines shapes please consult my PhD. thesis or article [1].



[1] L. Gisslen and R. Scholz, Submitted (2009).